滇黃精水提物促進羅伊氏乳桿菌生長增殖和定植的作用機制(二)
2方法
2.1滇黃精水提物的提取制備與菌株活化
參考陳秋定[4]方法提取滇黃精水提物及L.reuteri 1.2838的活化。
2.2滇黃精水提物對L.reuteri 1.2838增殖的影響
挑取活化后的單菌落接種于MRS培養液中培養至對數生長期作為種子液。將種子液按1∶100(v/v)接種量分別接種到MRS液體培養基和含有滇黃精水提物的MRS培養基(水提物濃度為0.0126 g·mL-1)中,37℃、180 r·min-1厭氧培養。自0 h起,每隔2 h吸取培養液加入96孔板中用全波長掃描多功能酶標儀測量OD600 nm值。所有組均設置3個重復,記錄數據,繪制生長曲線。
2.3滇黃精水提物對L.reuteri 1.2838群體感應信號分子AI-2生成的影響
2.3.1 L.reuteri 1.2838與滇黃精共孵育菌液的制備
向空白MRS培養液中添加滇黃精水提物至濃度為0.0126 g·mL-1,按1∶100(v/v)接種量接種2.1中活化的種子液。厭氧條件下,37℃、180 r·min-1振蕩培養16 h[4]。所得菌液在4℃條件下,12000 r·min-1離心10 min,棄去上清液,向所得菌體沉淀中加入磷酸鹽緩沖液(Phosphate buffer saline,PBS)重懸,再次以上述條件離心,棄去上清液,洗去滇黃精水提物殘留。向所得菌體中加入新的MRS培養液,調整菌液OD600 nm值為0.6-0.8,按1%接種量接種于新的MRS培養液中,37℃、180 r·min-1振蕩培養2 h,取培養后菌液2 mL,12000 r·min-1離心10 min取上清液用0.22μm的濾頭過濾,作為群體感應信號分子AI-2的待測樣品。空白對照為未添加滇黃精水提物共孵育的L.reuteri 1.2838菌液。
2.3.2自身誘導因子AI-2的檢測方法
參照陳秋定[4]和Ismail[9]的方法。制備Vibrio harveyi BB170菌株種子液,以1∶5000(體積比)轉接到2216E液體培養基中作為檢測樣品,取9 mL哈維氏菌檢測液加入2.3.1所得群體感應信號分子AI-2待測樣品1 mL,30℃、180 r·min-1通氣培養1 h。取2216E液體培養基重復以上操作,作為陰性對照組。到達培養時間后,每管吸取200μL加入96孔平底發光板中檢測,每個樣本設置3個復孔,用全波長掃描多功能酶標儀檢測其化學發光量,計算AI-2活性的相對光單位(Relative light unit,RLU)。
RLU=A÷B,式中,A——待測樣品化學發光量的平均值;B——陰性對照化學發光量的平均值。
2.4滇黃精水提物對L.reuteri 1.2838基因轉錄的影響
2.4.1 RNA的提取、文庫構建及測序
采用Trizol法提取L.reuteri 1.2838菌體RNA。利用超微量分光光度計對所提的RNA濃度和純度進行檢測,用Agilent 5300生物分析儀測定RNA質量。采用Illumina TruseqTM RNA sample prep Kit方法建庫。在Illumina NovaSeq 6000平臺進行轉錄組測序。
2.4.2轉錄組分析
實驗通過RSEM對基因表達水平進行分析,指標為FPKM(Fragments per kilobase of exon model per million mapped fragments),利用DESeq 2、Padjust等方面對基因的表達量及差異倍數進行統計分析,確定顯著差異表達基因,對差異基因進行GO分析和KEGG分析,確定差異基因的功能分布與涉及通路。
2.5逆轉錄實時熒光定量PCR驗證
為驗證轉錄組數據的準確性,實驗通過熒光定量PCR(Real-time polymerase chain reaction,RT-qPCR)驗證差異基因的表達水平。首先將2.4中獲得的RNA反轉錄成cDNA,再根據轉錄組測序分析的結果,以16S rRNA為內參基因,選擇dna K、ndk、secA基因進行驗證,通過Primer 5.0軟件設計引物,具體序列見表1。最后計算相對表達量差異2-ΔΔCT。
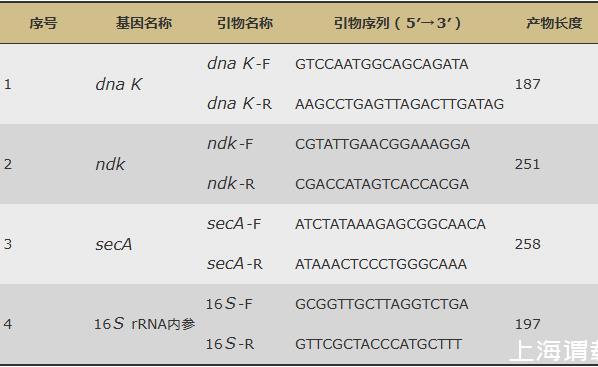
表1 qRT-RCR擴增特異引物
2.6數理統計與分析
所有實驗數據均進行3次重復,采用均值±標準差表示,采用GraphPad Prism 10統計軟件分析數據,進行T檢驗分析,檢驗結果均取*P<0.05,**P<0.01,***P<0.001,****P<0.0001作為統計學意義差異判斷標準。
相關新聞推薦
1、白馬耳組織成纖維細胞體外培養、冷凍前及復蘇后存活率、生長曲線繪制(二)
3、微生物生長動態監測系統的應用:研究β-內酰胺類抗生素對絲狀化的誘導能力